All You Need To Know About Aplastic Anemia Or Bone Marrow Failure (Hypoplastic Anemia)

Aplastic Anemia Patient
Introduction
This is anemia resulting from failure to produce the formed elements of blood by the bone marrow. Morphologically, in the vast majority of cases, the bone marrow is hypocellular or even totally acellular. Generally, all the elements are affected though at times the affection may be selective.
Aplastic anemia is a rare disorder contributing to 1-2% of the total number of anemia cases. Males suffer twice as many as females. The disease is primary (idiopathic) in over 75% of cases, in the rest, it is secondary to known casuses.
What Causes Aplastic Anemia?

Causes of Aplastic Anemia
Primary: Acquired idiopathic congenital (Fanconi’s)
Secondary: Chemicals and toxins including drugs
- Agents which regularly produce bone marrow hypoplasia
- Anti-cancer drugs (busulphan, cyclophosphamide, etc)
- Ionizing radiations
- Benzene and its derivatives
These damage the stem cells and arrest mitosis. If the damage is not severe, withdrawal of the drug is curative.
- Agents that occasionally produce bone marrow hypoplasia: Chloramphenicol, sulphonamides, phenylbutazone, gold, thiouracil, antoconvulsants, antidiabetic agents, insecticides, industrial solvents, hair dyes etc.
These are not dose related. Many have an immunological basis. Withdrawal of the drug may not improve the marrow aplasia.
- Infections -infective hepatitis (HAV), military tuberculosis
- Pregnancy
- Thymoma (autoimmune)
- Paroxysmal nocturnal hemoglobinuria (PNH)
- Systemic disorders such as chronic renal failure, hypothyroidism, and Addison’s disease may be accompanied by varying grades of marrow aplasia.
How Aplastic Anemia Came About
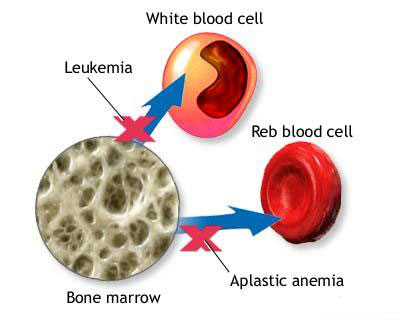

Pathophysiology
In this disorder, the proliferation and differentiation of hemopoietic stem cells are deranged. Most cases of aplastic anemia are due to a deficiency of marrow stem cells. Immune mechanisms may be responsible for bone marrow aplasia in some cases. A third suggested mechanisms is abnormality of the microenvironment in which the stem cells are proliferating.
The marrow is reduced in volume, often it is yellow due to increase in fat. It is difficult to obtaine a specimen and ‘dry taps’ are common. In an ordinary case, all the formed elements are reduced. Megakaryocytes, myeloid elements and erythroblasts are progressively wiped out resulting in thrombocytopenia, neutropenia and anemia as the disease evolves. In the final stage, all cells are uniformly reduced. Due to reduction in granulocytes, the lymphocytes, plasma cells and other reticuloendothelial cells may appear to be relatively increased.
How To Detect Aplastic Anemia
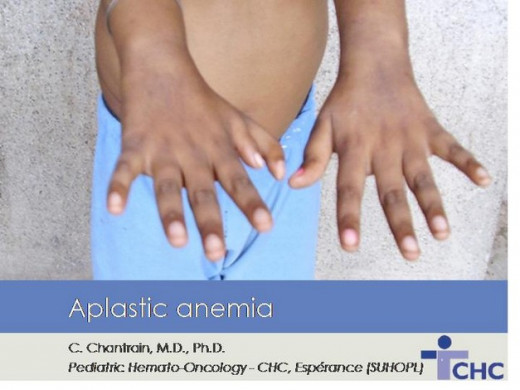
Clinical features
Symptoms are slow in onset, taking 1-2 months for manifestation. Sometimes bleeding and infection may set in early. Symptoms are due to the consequences of pancytopenia. Varying grades of anemia develop and often it is severe. Hemorrhagic manifestations occur due to thrombocytopenia. Common presentations are purpura, ecchymosis, epistaxis, bleeding from the gums, gastrointestinal bleeding and vaginal bleeding. Bacterial infections occur due to neutropenia. These present with fever or other localized infections.
Hematological features
The erythrocytes, granulocytes and platelets are all reduced. Reticulocytes may be absent. In some cases, granulocytes may be present in small numbers, but the young forms are considerably reduced. Erythrocytes are normocytic and normochromic with uniform size and shape. The bone marrow shows aplasia or hypoplasia. Stainable Iron in the marrow may be increased.
Managing An Aplastic Anemia Patient
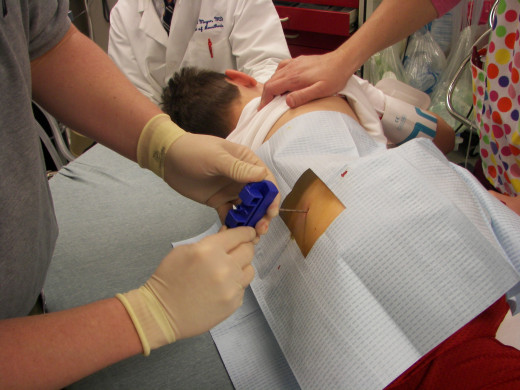
Diagnosis And Management
Diagnosis
The picture of peripheral pancytopenia with aplastic bone marrow confirms the diagnosis of aplastic anemia. Bone marrow biopsy is essential in all cases, since it helps to rule out conditions like acute leukemia, lymphomas, multiple myeloma, myelofibrosis, which may superficially resemble aplastic anemia. Peripheral pancytopenia with normocellular or hypercellular marrow should suggest the possibility of aleukemic leukemia, sideroblastic anemia, dyserythropoietic states, and paroxysmal nocturnal hemoglobinuria. Acute myeloid leukemia may be mistaken for alastic anemia in the initial stages, but presence of splenomegaly is suggestive.
Thrombocytopenic purpura, agranulocytosis and hemolytic anemia with aplastic crises cause difficulty in diagnosis at times. In immune thrombocytopenia, the marrow shows increase in the number of megakaryocytes. In agranulocytosis, only the granulocyte precursors are diminished. Other elements are normal. In hemolytic anemia with aplastic crises, erythroid precursors are selectively depressed.
Course and prognosis
The course is variable in aplastic anemia and the course may extend for periods ranging from a few months to even years. Severe cases have particularly grave prognosis and the median survival is less than 4 months and 80% die within 1-2 years if untreated. Early response to therapy is a favourable sign. About 80% of nonresponding patients die within 3-4 months. Life can be prolonged for several years by proper substitution therapy.
Management
This consists of
- Withdrawal of the offending agents;
- Supportive measures to compensate for the pancytopenia and prevent complications; and
- Positive attempts to restore bone marrow activity.
Supportive measures include transfusion of blood or packed red cells, platelets and granulocytes. The patient should be isolated and care should be taken to prevent cross infection from attendants and endogenous infections by the commensal flora. Infection should be diagnosed early and treated with bactericidal antibiotics.
© 2014 Funom Theophilus Makama




