HYPOPITUITARISM-DIAGNOSTIC CRITERIA AND TREATMENT (Growth Hormone deficiency and Pituitary dwarfism)

THE HYPOTHALAMIC – PITUITARY RELATIONSHIPS
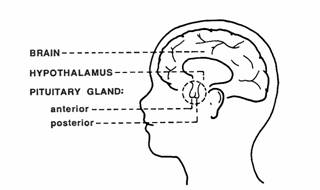
HYPOTHALAMUS
The hypothalamus modulates the activity of the pituitary gland (hypophysis) via 2 distinct routes, one to the posterior and other to the anterior pituitary. The hypothalamus is connected with the posterior pituitary (neurohypophysis) by the supraopticohypophyseal nerve tract (peptidergic neurons). No nervous connection with the anterior pituitary (adenohypophysis) exists, but blood coursing through the hypothalamus gathers into a portal venous system which traverses the anterior pituitary and thus serves as a channel for direct transmission of the hypothalamic neurohormones. By these 2 routs, the hypothalamus is able to stimulate or release pituitary hormones.
Neurohypophyseal function.
The supraoptic and paraventricular nuclei of the hypothalamus secrete 2 octapeptide hormones – vasopressin and oxytocin. Both hormones are transported down axons bound as granules to carrier proteins, and are stored in the nerve endings of the posterior pituitary. They are released from these endings into the general circulation, where the granules dissociate, releasing free hormone and proteins.
Vasopressin (antidiuretic hormone, ADH) regulates water balance by stimulating resorption in the distal renal tubule. Secretion of ADH is controlled by increased osmotic pressure and decreased effective plasma volume.
Oxytocin stimulates uterine contractions (more so as pregnancy progresses), causes the myoepithelial cells of the breast to contract, expressing milk into the ducts. Secretion of oxytocin is controlled by suckling.
Adenohypophyseal function.
The hypothalamus plays an important role in hormone regulation by secreting a series of small peptides which stimulate or inhibit the synthesis and release of hormones by the anterior pituitary. Traditionally, these hypothalamic peptides have been known as releasing factors or ashypophysiotropic hormones (because they affect not only hormone release but also secretion). The neurons that secrete the hypothalamic peptides are located in the ventromedial nucleus nearby the median eminence. Secretion of these peptides is intimately related to the release of neurotransmitters by neurosecretory cells which are located in the same anatomical area (the best known neurotransmitters are the catecholamines dopamine and norepinephrine and the indoleamine serotonin).
The corticotropin-releasing hormone (CRH) stimulates the synthesis and release of corticotropin (ACTH) by the anterior pituitary.
The thyrotropin-releasing hormone (TRH) controls the secretion of thyrotropin (TSH) and also causes release of prolactin.
The gonadotropin-releasing hormone (GnRH), or luteinizing hormone-releasing hormone (LHRH) stimulates pituitary secretion of luteinizing hormone (LH) and to lesser extent that of follicle-stimulating hormone (FSH).
The growth-hormone-releasing hormone (GRH) stimulates and growth-hormone-inhibiting hormone (GIH, or somatotropin release-inhibiting factor (SRIF), or somatostatin) inhibits growth hormone (somatotropin) secretion from the anterior pituitary. GIH also inhibits insulin and glucagon secretion, gastrointestinal secretion of water, bicarbonate, pancreatic enzymes, gastric acid gastrin; glucose absorption and calcium transport.
The prolactin-inhibiting factor (PIF) controls prolactin secretion.

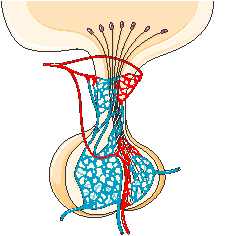


PITUITARY GLAND
The pituitary gland lies in a bony structure, the sella turcica, located at the base of the skull. The gland is a small organ about I cm long; it weighs 500 mg and is divided into two parts, anterior (adenohypophysis) and posterior (neurohypophysis). Both parts derive from the ectoderm, but their origins differ. The anterior pituitary originates from Rathke's pouch, an evagination from the roof of the embryonic oral region, whereas the posterior pituitary is formed from an extension of the floor of the third ventricle. Secretory granules can be demonstrated in the fetal pituitary by the end of the 12th week. The blood supply reaches the anterior pituitary through the superior hypophyseal artery (a branch or the internal carotid) and through the venous portal system, which originates in the median eminence and ends in the sinusoidal capillaries of the anterior lobe. Hypothalamic peptides reach the adenohypophysis by way of the portal system. Thin nerve fibers from the carotid plexus surround the arterioles.
The anterior pituitary secretes several important hormones, some of which in turn control hormone secretion by other glands: they are corticotropin (ACTH), somatotropin (growth hormone), prolactin, gonadotropins [follicle-stimulating (FSH) and luteinizing (LH) hormones], thyrotropin (TSH), and melanocyte-stimulating hormones (MSH).
FEEDBACK : Hormone secretion → delivery to target cells → hormone recognition by receptors in target cells → biologic effect → hormone degradation → signal from target cells to slop further hormone secretion .
ACTH stimulates adrenal growth and the secretion of cortisol. ACTH secretion is under dual control: by the hypothalamus through CRH (stress provokes serotonin secretion, and last stimulates secretion of GRH) and by adrenals through cortisol (by negative feedback). ACTH is secreted episodically, in a circadian pattern, with higher plasma concentrations in the early morning hours.
Somatotropin (growth hormone) stimulates protein synthesis and lipolysis, and increases blood glucose concentration (by decreasing of glucose utilization by the peripheral tissues and stimulating of hepatic gluconeogenesis). Somatotropin is under the control of GRH and GIH.
Prolactin function is initiation and maintenance of lactation. (Before pregnancy, the development and differentiation of the breast are influenced by estrogen, progesterone, and prolactin, but steroids are not effective in the absence of prolactin). Prolactin is under the control of TRH, PIF and physiologic stimulus for prolactin secretion is suckling.
Gonadotropins: FSH stimulates follicular growth in the female and spermatogenesis in the male;LH induces ovulation and maintains the corpus luteum after ovulation has occurred in the female and stimulates testosterone secretion by the Leydig cells of the testes. Gonadotropins secretion is under dual control from the hypothalamus (GnRH production is stimulated by norepinephrine and inhibited by dopamine) and the gonads (feedback mechanism).
TSH stimulates the vascularity of the gland and the hypertrophy of the follicular cells, it also has extrathyroid actions, e.g. stimulation of lipolysis. TSH secretion is under the control of TRH and the thyroid hormones (feedback mechanism).
MSH stimulates hyperpigmentation. MSH secretion is under the control of ACTH.
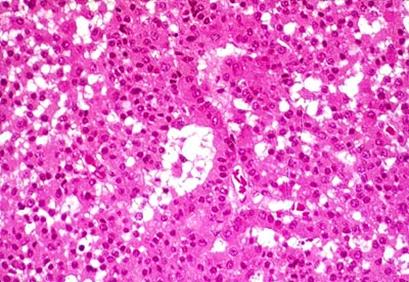

 with the infarcted necrotic area (on the left). Notice the ghost architecture and the acellular area of infarction in this case of Sheehan'ssyndrome.")


, magneto resonance investigation (MRI (pict.)) and cerebral angiography. P")

HYPOPITUITARISM
It is the syndrome, which is characterized by deficiency of one or more anterior pituitary hormones.
Etiology
Several pathologic processes may play an etiologic role in the development of the; syndrome:
1. Tumors:
- chromophobe adenomas can cause hypopituitarism by destroying the anterior pituitary cells that secrete the trophic hormones. Hormone deficiencies in order of frequency are growth-hormone, gonadotropins, TSH, ACTH, and prolaclin;
- craniopharyngiomas arise from remnants of Rathke's pouch and can be either cystic or solid. This tumor is more common in younger people (with peak incidence in the second decade) who present with signs and symptoms of increased intracranial pressure; visual field defects, particularly bitemporal hemianopsia; hypogonadism; growth retardation; diabetes insipidus; and suprasellar calcifications;
- pituitary cysts are very common, with a frequency as high as 25 % in autopsy series. The cysts vary considerably in size. Some produce little change in the sella, while others may show moderate supraseliar extension or even massive sellar erosion.
2. Ischemia:
- postpartum uterine hemorrhage (Sheehan's syndrome) is the most common cause of hypopituitarism in females. The clinical manifestations result from pituitary necrosis, which is believed to be caused by ischemia. Anterior pituitary hormone deficiencies appear during the puerperium. The neurohvpophysis is seldom affected. Failure to lactate and involution of the breasts are followed by loss of axillary and pubic hair. The amenorrhea of pregnancy persists and the symptoms of TSH and ACTH deficiency appear later. Although gonadotropins and growth-hormone deficiency may be the earliest to occur, there is variability in the order in which the hormones fail and in the type of hormones. affected.
- less frequently, involvement of the vascular supply to the anterior piluitary in patients with diabetes mellitus, sickle-cell disease, and collagen vascular diseases can result in the development of pituitary insufficiency.
3. Many cases of hypopiluilarism that were considered idiopalhic probably represent an autoimmuneprocess in which antipituiatary antibodies are present. Lymphocytic hypophysitis is a syndrome reported in women only during pregnancy or the postpartum period. Signs and symptoms resemble those seen in patients with pituitary tumors. Light microscopic studies reveal extensive cellular infiltration of the pituitary by lymphocytes. Antipituitary antibodies, particularly to prolactin, have been detected in some of the patients. The entity is considered to be autoimmune and it is associated with other endocrine autoimmune processes, notably chronic thyroiditis.
4. Hypopituitarism frequently results from pituitary irradiation. Eighty-three % of patients who receive radiotherapy for tumors of the head and neck, usually 5000 rads, develop pituitary insufficiency. Hormone deficiencies may develop within a year or more after irradiation. The hypothalamus, the pituitary, or both may be affected.
5. Infiltration of the pituitary gland by iron, histiocytes, or granulomatous processes is a relatively rare cause of hypopituitarism:
- hemochromatosis;
- histiocytosis;
- sarcoidosis.
6. Infectious processes:
- tuberculosis;
- bacteria;
- fungus.
7. Pituitary insufficiency can be caused by traumatic or surgical destruction of the hypothalamic-pituitary area.
Clinical presentation.
Many patients with pituitary tumors are asymptomatic, and the diagnosis is made accidentally by plain x-rays of the skull obtained. Usually, clinical manifestations of hypopituitarism are not present unless 75 % of the gland is destroyed.
Some patients present with signs and symptoms of increased intracranial pressure, such asheadaches, vomiting, and papilledema. Compression of the optic chiasm results in visual impairment, notably bitemporal hemianopsia.
In the classic case of hypopituitarism, clinical manifestations of ACTH, TSH, and gonadotropin deficiency are presen:
1. Weakness, dizziness, weight loss, and abdominal symptoms are secondary to decreased corlisol secretion by the adrenals. Symptoms of hypoglycemia may predominate. Dehydration, hypotension, and prostration may be found on physical examination.
2. Gonadotropin deficiency is responsible for decreased libido in patients of both sexes and for amenorrhea in females. Decreased libido, impotence, and hair loss are caused by testosterone deficiency, and amenorrhea is caused by estrogen deficiency. In males, a sallow complexion of the face, prominent wrinkles of the forehead, and loss of body hair, particularly pubic and axillary hair, may be very striking.
3. Patients with unexplained anemia should be investigated for hypopituitarism. Erythropoiesis is hormone-dependent, and is particularly dependent upon testosterone, which stimulates erythropoietin production by the kidney.
4. Secondary hypothyroidism is characterized by tiredness, cold intolerance, constipation, tingling and numbness of the extremities, and anorexia. Pale, dry skin and slow relaxation phase of the deep tendon reflexes are observed on examintion of the patient.
The symptoms of hypopituitarism mimic those of many diseases. The diagnosis can often be missed unless it is suspected. Watch for pituitary insufficiency:
1. In women who fail to lactate following pregnancy and delivery. (Involution of breasts and sparse axillary and pubic hair would suggest the diagnosis.)
2. In females with unexplained arnenorrhea, particularly if symptoms of ACTH and TSH deficiency or increased intracranial pressure are present.
3. In males with decreased libido and loss of axillary and pubic hair.
4. In patients with unexplained anemia.
5. In diabetic patients who experience a reduction in insulin requirements.
Diagnosis.
1. The presence of pituitary insufficiency should be suspected from the history and physical examination and confirmed by laboralory lesling. There are two types of diagnostic procedures: those designed to identify the cause of the problem (usually a radiologic procedure) and those intended for evaluation of pituitary function.
2. Laboratory findings:
- anemia, hypercholesterolemia, tendency to hypoglycemia;
- hormonal assessment (decreased levels of pituitary and peripheral endocrine glands).
3. Instrumental investigations.
X-ray diagnosis of the abnormal sella turcica.
The sella turcica can be evaluated by plain x-ray films of the skull, multidirectional polytomography, pneumoencephalography, computerized cranial axial tomography (CT scan),magneto – resonance investigation (MRI (pict.)) and cerebral angiography. Plain films of the sella are useful in evaluating patients for the possibility of a pituitary tumor. The sella may be abnormal in size, volume, or configuration. The upper limits of normal for sellar dimensions are length 17mm, depth 13mm, and width 15mm. Sellar enlargement is seen in 90 % of patients with endocrine deficiency caused by a pituitary tumor, but it may also be seen in patients with increased intracranial pressure, the empty sella syndrome, internal carotid artery aneurysm, primary hypothyroidism, and juxtasellar tumors. Likewise, a normal sella does not rule out pituitary pathology because microadenomas which are less than 1 cm in diameter may not be visualized.
Cerebral angiography is useful in establishing the presence of parasellar lesions, determining the degree of suprasellar or parasellar extension of a pituitary tumor, excluding the presence of an aneurysm, and determining whether major vessels are involved by the tumor.
Visual fields. A search for visual-field defects is important. Visual impairment in patients with hypopituitarism suggests the presence of a pituitary tumor compressing the optic chiasm. Bitemporal hemianopsia is the typical finding but other visual-field defects may be present. This ophthalmologic procedure can also help to monitor the response to therapy and to evaluate patients for possible tumor recurrences.
Treatment
The treatment of patients with pituitary insufficiency consists of
1) eliminating the underlying cause and
2) replacing the deficient hormones.
Pituitary tumors should be removed surgically, although irradiation and drug therapy (bromocriptine) are also available.
Hypothalamic peptides or pituitary hormones are not suitable for hormone replacement for several reasons:
(1) The human hormones are difficult to oblain in pure form;
(2) because of their nature and short halh-life they have to be given parenterally and frequently; and
(3) since they stimulate antibody formation, their activity is lost a few weeks after initiation of therapy.
Under these circumstances the usual practice is to administer the hormones produced by the target glands. They are available in pure form and are relatively inexpensive.
Adrenal insufficiency. Hydrocortisone is given orally in a total daily dose of 20 to 30 mg. Many physicians give one 20-mg tablet in the morning and half a tablet in the afternoon to try to stimulate the normal diurnal variation of plasma cortisol. This practice is not necessary, the local amount can be given as a -single dose in the morning. Other steroids with glucocorticoid activity can be used. Prednisone is preferred by some physicians because of its lower cost.
Hypothyroidism. Symptoms are easily controlled with synthetic l.-thyroxine. Most patients need 0.15 mg/day. In women the replacement dose is usually lower, and many do well on 0.1 mg/day.
Hypogonadism replacement with gonodal steroids is never indicated until puberty normally occurs. These agents in high doses can hasten bone maturation and epiphyseal closure, thereby limiting the height which may ultimately be reached.
In males testosterone therapy is recommended. (Testosterone enanthate, a long-acting preparation at a dose which is equiv alent to 50 to 100 mg/week. This is given as an intramuscular injection of 100 to 200 mg every 2 to 4 weeks. At the beginning of therapy, it is useful to administer 200 mg IM every 2 weeks until a good androgenic response is obtained. Side effects of testosterone include gynecomastia, acne, and occasionally hypertension, presumably secondary to stimulation of sodium retention by testosterone. The indications for testosterone therapy include decreased libido and anemia).Methyltestosterone or fluoxymesterone (10 to 40 mg/day) can also be used. They have the advantage of convenience, since they can be given orally, but development of cholesiatic jaundice and liver disease are potential problems with their use.
Premenupausal females with ovarian failure should be treated with estrogens. It can be ethinyl estradiol, which is administered orally in a dose of 0,025 to 0,05 mg once a day for 25 days of each month. Although this is the usual replacement dose. some patients may need a smaller or a larger dose. The benefits of estrogen therapy include return of menstruation, improvement of secondary sex characteristics such as skin texture and breast development, increased libido, and prevention of osteoporosis.
Side effects. Estrogen therapy is not without risk, e.g., the increased incidence of thrombophlebitis and pulmonary embolus. Hypertension, glucose intolerance, liver disease and hyperlipoproteinemia are also potential problems. There is also some controversial evidence that estrogens predispose to carcinoma of the endometrium, To minimize all these side effects, it is recommended that the minimum effective estrogen dose be used, that estrogens be prescribed cyclically rather than on an everyday basis, that periodic pelvic examinations be performed, and that an endometrial biopsy be done at the first sign of unexpected uterine bleeding.







 Because of the slow progression of the disease, the sella is usually enlarged when the patient is first seen.")
 can reduce the secretion of growth hormone without causing hypopituitarism in 88")
GROWTH-HORMONE EXCESS
Increased secretion of growth hormone (GH) by pituitary tumors or increased sensitivity of peripheral tissues to GH leads to gigantism before puberty and to acromegaly after puberty.
Etiology
1. Acidophilic adenoma of the pituitary.
2. Chromophobe adenoma of the pituitary
3. Ectopic GH-producing tumors. The signs and symptoms which appear in these patients are of two types:
1) those secondary to the presence of tumor: mass effect (Headaches and visual impairment are present in 90 and 60 % of the patients respectively. Typically one finds bilateral loss of peripheral vision (bitemporal hemianopsia), which may be progressive.) and
2) those secondary to increased circulating growth-hormone levels.
GIGANTISM
is characterized by height more than 190 cm in women and 200 cm in men. Hypersecretion of GH prior to closure of epiphysis leads to proportional growth of bone; both length and width of bone are increased.
Differential diagnosis have to be made with
- constitutional high height
- Klainfelter’s syndrome
- Marphan syndrome
ACROMEGALY
GH hypersecretion after closure of epiphyses leads to periostal overgrowth and cortical thickening. Overgrowth of the mandible leads to protrusion of the jaw (prognatism). There is an overbite and the teeth become separated (diastema). Bone overgrowth and soft tissue thickening lead to characteristic coarsening of the facial features. The hands are widened and the fingers become broad, requiring a larger ring size. Similiar changes in the feet require a larger shoe size. This increase in dimension of the acral (distal) parts of the body has led to the term acromegaly.
Hepatomegaly and cardiomegaly are consistent findings. Thyromegaly is present in about 25 % of the patients. Hyper- and hypothyroidism occurs rarely. The kidney may also become enlarged, and renal clearance of phosphate is frequently impaired.
Hypertension is present in 40 % of patients with acromegaly, although its cause is unknown. It is unclear whether the increase in blood pressure is caused by growth hormone per se or whether it is part of the generalized cardiovascular involvement seen in patients with the disease. Plasma renin may be high, normal, or low. The association of primary aldosteronism with acromegaly has been reported in a few cases.
Other manifestations of the disease are secondary to the abnormalities in glucose metabolism which result from growth-hormone excess. Thus, symptoms of diabetes mellitus, which include polyuria, polydipsia, polyphagia, and tiredness, may be present.
Increased perspiration is common among 90 % of patients. The skin is thickened, and females may note hypertrichosis.
Joint manifestations are common (70 %) in patients with acromegaly. In some patients, for example, arthralgias and arthritis may be the presenting complaints.
Amenorrhea, which occurs in as many as two-thirds of female patients, may be the chief complaint. Decreased libido is experienced by one-third of the patients.
Hyperfunction or deficiency of other pituitary trophic hormones may be present. Of note is the frequency among women with acromegaly (15 % of cases) of galactorrhea which is caused by increased secretion of prolactin by the pituitary tumor.
Diagnosis
The diagnosis of acromegaly is usually suspected from:
1) the patient's history and
2) physical examination (The head is large, the mandible is prominent, and the tongue is enlarged. The skin is thick and hard. Large hands and feet and increased subcutaneous tissue are evident on examination. Carpal tunnel syndrome occurs in as many as 44 % of the patients. Deformed joints can be confused with other arthritic syndromes.).
3) The role of the laboratory is to assist in the confirmation of the diagnosis. Since the syndrome is caused by excessive growth-hormone production, one wants to demonstrate increased circulating levels of this hormone. An occasional blood sample drawn at 8 a.m. may be sufficient if the values are grossly elevated. Unfortunately, this procedure is not very reliable for two reasons:
- some patients with acromegaly have normal growth-hormone concentrations, and
- in normal subjects growth-hormone levels vary greatly. Stress, sleep, exercise, food ingestion, and diurnal variation can significantly affect growth-hormone concentrations. Thus, a normal serum growth-hormone level does not exclude acromegaly, and an elevated growth-hormone level does not always mean acromegaly.
To eliminate this problem one takes advantage of the concepts of feedback and autonomy to screen patients for acromegaly. Normally, glucose suppresses growth-hormone secretion. In patients with acromegaly the pituitary tumor is autonomous; that is, it does not respond to hyperglycemia. The recommended procedure consists of a glucose tolerance test with concomitant growth-hormone determinations; serum growth-hormone concentration falls in normal subjects 1 h after the oral glucose load. Patients with acromegaly not only have elevated values but also fail to suppress in response to glucose ingestion.
4) Somatomedin-C, the tissue mediator of some of the effects of growth hor mone, has been measured by radioimmunoassay in the blood of patients with acromegaly. Because somalomedin-C binds to plasma protein, its serum concentrations are more stable than those of growth hormone. The values are evaluated in patients with acromegaly and they correlate well with growth hormone values 1 h post glucose administration.
5) A paradoxical growth-hormone increase in response to TRH is seen in some patients with acromegaly, and when this abnormality is found, it can be used to follow the response to therapy.
6) Glucose intolerance is seen in about 40 % of the patients. This information becomes available at the time the glucose tolerance test is ordered to obtain growth-hormone levels.
7) An important part of the evaluation in patients with acromegaly is the search for visual-field defects. As mentioned earlier, because of the anatomical location of the tumor, the typical finding is the presence of bitemporal hemianopsia. However, other visual-field defects may be present.
8) The sella turcica is enlarged in at least 90 % of patients with acromegaly. (MRI , CT) Because of the slow progression of the disease, the sella is usually enlarged when the patient is first seen.
9) One should also be interested in determining whether suprasellar extension of the tumor is present, and the best procedure for this purpose is computed tomography. This information can help the surgeon to decide what approach to use when removing the tumor.
Treatment.
Three therapeutic modalities—surgery, irradiation, and drugs—have been used to treat patients with acromegaly.
The surgical techniques of craniotomy, transsphenoidal hypophysectomy, and stereotaxic surgery, particularly cryosurgery, have been used to treat acromegaly, and all of them have met with some success.
At present, transsphenoidal hypophysectomy is the procedure of choice. An incision is made in the inner aspect of the upper lip and the pituitary gland is entered through the nasal septum and the sphenoid sinus. With this procedure, improvement or cure of the acromegaly can be achieved in nearly 90 % of the patients. Advantages of transsphenoidal hypophysectomy, besides its effectiveness, include simplicity and low morbidity. Hypupituitarism and diabetes insipidus are extremely uncommon, since the re mainder of the pituitary gland is left intact. Perhaps the major difficulty of transsphenoidal hypophysectomy is the fact that some patients need a second operation because of recurrence of symptoms.
Craniotomy is reserved for large tumors with suprasellar extension and involvement of the optic chiasm. Cryohypophysectomy (destruction of the pituitary by cold injury) can reduce the secretion of growth hormone without causing hypopituitarism in 88 % of the patients.
External irradiation has been reported by some to control the activity of the disease in as many as 80 % of the patients, but other reports indicate that the procedure has not been too successful. One of the limitations is the amount of radioactivity that can be delivered, which should not exceed 5500 roentgens (R). Another difficulty with irradiation is its slow onset of action, the full effects requiring as long as 10 years to appear.
Irradiation with accelerated proton beams, which can be focused on the pituitary, delivers 10,000 or more rads without damaging the surrounding tissues. Results with this technique and those obtained with alpha particle irradiation are similar.
Drugs.
A dopaminergic agonist and ergot derivative, 2a-bromergocriptine (bromocriptine), causes a paradoxical inhibition of growth-hormone secretion in patients with acromegaly. In doses of 10 to 60 mg/day, the drug has induced clinical remissions in 73 % of the treated patients. However, return of normal growth-hormone suppressibility in response to glucose is seen in only 22 % of patients. Side effects include nausea, orthostatic hypotension, consti pation, digital vasospasm, and peptic ulcer.


Contributors







PITUITARY DWARFISM (GROWTH FAILURE)
it is the disease caused by decreased secretion of GH by pituiatary gland or decreased sensitivity of peripheral tissues to this hormone and leads to growth retardation.
The height of adult men is less than 130 cm, and adult women is less than 120 cm.
Etiology
Children with pituitary dwarfism most commonly have either a craniopharyngioma or no demonstratable etiology (idiopathic hypopituitarism). The latter is more frequent in males than in females. Before the diagnosis of pituitary dwarfism can be made, all other etiologies must be ruled out (look differential diagnosis).
Classification.
A. Organic (trauma, neoplasms, infection).
B. Idiopatic (primary or secondary, due to hypothalamic deficiency).
1. Panhypopituitarism.
2. Isolated GH deficiency (may be hereditary and transmitted as an autosomal recessive trait, in other instances a hereditary basis cannot be established).
Clinical manifestations
1. Child is born with normal weight and height.
2. Growth retardation can be observed since 3 – 4 years (the increasing of the height is not more than 1 cm per year.
3. The patient, despite small size, has normal body proportions.
4. Mental develoment is normal.
5. Secondary hypothyroidism.
6. Secondary adrenal insufficiency.
7. Puberty will not appear because of a lack of gonadotropic hormons (secondary hypogonadism).
8. The passport age is not corresponding with biologic age.
9. In patients with isolated GH deficiency the patients have normal pituitary function (other than lack og GH), undergo normal puberty, and have normal reproductive capacity.
Diagnosis
Clinical picture (carefully recorded growth charts may disclose the time of onset of the disease); level of GH; X-ray examination of the skull and hands can be helpful in diagnosis.
Differential diagnosis have to be made with other causes of short stature:
I. Endocrine:
1. Primordial (Lorrain – Levi) dwarfism (is an ill-defined diagnosis which presupposes normal hormonal activity in an individual with limited potential for tissue growth. Because the infants in this category are often small for gestation age, the term intrauterine dwarfism has also been used to designate those who have growth retardation at birth. While patients with hypopituitarism are normal of size at birth (probably because of maternal Ghand somatomedin), patients with premordial dwarfism are already dwarfed at thus time because maternal somatomedin is not available or is ineffective. Characteristically, growth is slow from earliest infancy, although epiphyseal maturation and sexual development occur normally or at only a slightly retarded rate. No definite abnormalies have been reported. Intellect may be affected and other congenital anomalies may be present.)
2. Hypothyroidism (primary or secondary)
3. Precocious puberty.
4. Cushing’s syndrome.
II. Constitutional (normal variant) short stature .
III. Nonendocrine disorders (such as chronic disease, malabsorption syndromes, certain hematologic diseases, diseases involving the skeletal system are also common causes of growth retardation).
IV. Psychosocial dwarfism (is associated with severe emotional deprivation, and is alleviated by the removal of the child from the adverse situation).
Treatment.
I. Balanced diet.
II. Complex of physical exercises.
III. Pharmacotherapy.
1. GH (synthetic). Indications: a low level of GH or a low level of somatomedin in patients with biologic age less than 12 – 13 years and passport age more than 3 years. Doses of 2 – 4 IU of GH given IM at nighttime three times a week for 2 – 3 months with 2 – 3 months break have accelerated the rate of linear growth, through often this therapeutic effect is not sustained.
2. Anabolic steroids under the control of biologic (osteal) age.
3. Thyroid replacement.
4. Replacement with gonodal steroids is never indicated until puberty normally occurs. These agents in high doses can hasten bone maturation and epiphyseal closure, thereby limiting the height which may ultimately be reached.
5. Vitamintherapy.
IV. Surgical therapy (a craniopharyngioma presents special therapeutic problems, usually necessitating removal of tumor tissue or drainage of fluid from tumor cysts.
References
1. The Merck Manual of Diagnosis and Therapy (fourteenth Edition)/ Robert Berkow and others. – published by Merck Sharp & Dohme Research Laboratories, 1982. – P. 987 – 992, 916 – 921. 990 – 997. 1032 – 1037, 1680 – 1681.
2. Endocrinology (A Logical Approach for Clinicians (Second Edition)). William Jubiz.-New York: WC Graw-Hill Book, 1985. - P. 21 – 33. 34 – 38,52 – 63. 416 - 497.
3. Manual of Endocrinology and Metabolism (Second Edition)/ Norman Lavin. – Little, Brown and Company.- Boston-New York-Toronto-London, 1994. - P. 55 – 56. 179 - 274.







