Porphyrias: Clinical Classifications, Physical Presentations, Diagnosis And Treatment

Clinical Presentations Of Porphyrias
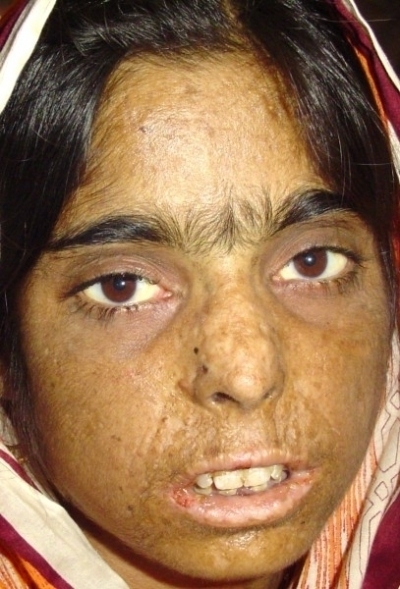
A General Overview
These are rare metabolic errors involving enzymes concered with the heme synthetic pathway. Most of these disorders are inherited, but porphyria cutanea tarda can be acquired. The synthesis of heme involves 8 steps and each step is enzymatically mediated. These are:
Glycine--------- Delta-aminolevulinic acid------------------ Porphobilinogen--------------- Polypyrrole------------- Uroporphyrinogen III------------------ Coproporphyrinogen III------------ Protoporphyrinogen III--------------------- Protoporphyrin III--------------- Heme.
In prophyrias, the skin and nervous system are predominantly involved. Excess porphyrins in the skin induce photosensitization and cause further damage. The pathogenesis of neural lesions is not fully understood. Porphyrias can be classified in different ways.
- Depending on the organ in which excess of metabolites are produced
- Clinical manifestations, and
- Specific enzyme defects
Clinical classification:
- Cutaneous manifestation without neurological features: Congenital erythropoietic porphyria, erythrohepatic porphyria (erythropoietic protoporphyria), porphyria cutanea tarda.
- Neurological features without skin lesions: Acute intermittent porphyria.
- Those that produce either cutaneous or neurological disease: Varigate porphyria (Porphyria varigata), hereditary coproporphyria.
Among the porphyrias, the most commonly encountered one is acute intermittent porphyria.
Signs Of Acute Intermittent Porphyria
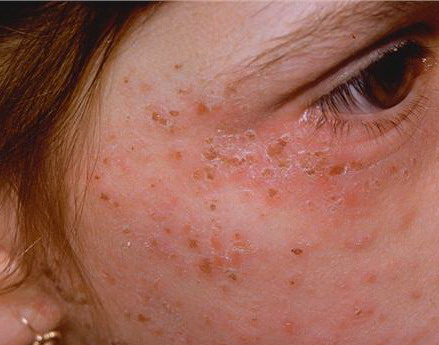
Diseases

Acute Intermittent Porphyria
It occurs worldwide. It is inherited as autosomal dominant. Women suffer more than men. Basic defect is a gross reduction (below 50%) of uroporphyrinogen I synthase which is required for the conversion of porphobilinogen to polypyrole. Urine contains the porphyrin precursors porphobilinogen (PBG) and delta-aminolevulinic acid (ALA) even when the disease is asymptomatic. During acute attacks, these metabolites increase in amounts. In addition, urinary uro and coproporphyrins also may increase.
Clinical features: The condition may remain latent for long periods without recognition. Acute episodes are precipitated by several factors. Important among them are:
- Drugs- barbiturate, sulpha drugs, griseofulvin, phenytoin, female sex hormones and several others.
- Dietary factors- starvation or excess eating; and
- Infections.
The acute attack is characterized by neurological phenomena and acute abdominal pain. Excitement, behavioural disturbances, depression, suicidal tendency, delirium, convulsions and coma are frequent. Peripheral neuropathy and mononeuropathies may be prominent in some cases. Autonomic neuropathy results in gastrointestinal dysfunction, urinary retention, hypertensive crises, disordered sweating and tachycardia.
Abdominal pain may be so severe as to be mistaken for a surgical emergency. Severe constipation or diarrhea may occur. Gastrointestinal fluid losses lead to hyponatremia and dehydration.
Diagnosis: Acute intermittent porphyria should be considered in the differential diagnosis of all bizarre neurological and neuropsychiatric disorders, severe abdominal pain and autonomic disturbances. Diagnosis is established by urine examination. Freshly passed urine is colourless but on standing for a few hours at room temperature, it becomes portwine in colour and this should be looked for in all cases. Porphobilinogen (PBG) in urine can be demonstrated by Watson- Schwartz test i.e. the urine gives a pink colour with Ehrlich’s aldehyde reagent just as in the case of urobilinogen. On extracting the pigment with chloroform, PBG is not extractable, whereas urobilinogen is. The amount of PBG can be estimated quantitatively.
Apart from acute intermittent porphyria, PBG may occur in urine in hepatic diseases, drug toxicity, porphyria variegata and hereditary coproporphyria. Specific diagnostic test is the demonstration of diminution of uroporphyrin I synthetase in erythrocytes.
Treatment- Symptomatic Management during an attack: The offending drugs should be stopped. Phenothiazine (chlorpromazine) administered in a dose of 50 mg intramuscularly relieves abdominal pain and the hypertension. Maintenance of fluid and electrolyte balance helps to tide over the crisis. High carbohydrate intake and propranolol give symptomatic relief. Aspirin, morphine, guanethidine, reserpine and penicillin can be safely used for suitable indications.
Prophylaxis should be in the form of avoidance of precipitating factors. Relatives should be screen for the enzyme deficiency and susceptible persons should be warned to avoid the offending drugs.
© 2014 Funom Theophilus Makama
Related

Kidney Pain: Location, Diagnosis and Treatment
 or Premature Ovarian Failure (Pof)")
Primary Ovarian Insufficiency (Poi) or Premature Ovarian Failure (Pof)

Night Sweats -Causes, Diagnosis and Treatment

Primary Peritoneal Cancer: Symptoms, Diagnosis, Treatment, and Clinical Trials

Mystery Diagnosis: Rsd - Reflex Sympathetic Dystrophy and Crps - Complex Regional Pain Syndrome


