What is Cystic Fibrosis and How is it Inherited?



What is Cystic Fibrosis? Animation Video
The movement of salt and water through cells
What is Cystic fibrosis
“Yesterday is history, tomorrow is a mystery, today is a gift of God, which is why we call it the present.”
(Bill Keane)
My introduction to the awful condition that is Cystic fibrosis came when I was a young newly qualified nurse working on the children's ward at the Brompton Hospital in Chelsea. The courage and bravery of the children who came to Rose Ward were one of the reasons why I decided to become a critical care paediatric nurse.
While caring for sick children proved to be exceedingly rewarding and fulfilling, it was also heart-rending to witness the suffering and extreme discomfort the children suffered, but admirable, they soldiered on. The Brompton, now the Royal Brompton Hospital is the largest specialist heart and lung center in the UK and one of the largest in Europe, so, of course, we saw many children with Cystic Fibrosis in varying degrees of severity. Working with children in this situation is tough, even for the seasoned professionals.
Cystic Fibrosis
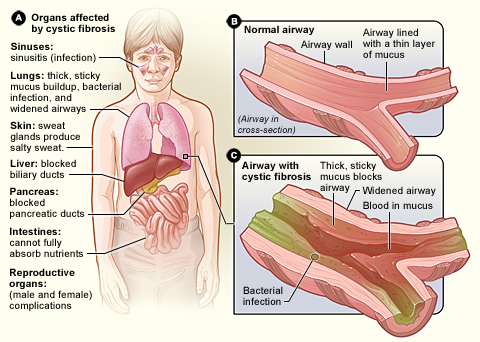
Cystic fibrosis (CF) is a genetic disorder caused by a defective gene known as the CFTR gene (Cystic Fibrosis Transmembrane Conductance Regulator). CF is a life-shortening inherited disease. The condition occurs when a faulty gene is passed from parents to a child causing the lungs, pancreas and digestive system to become clogged with thick, sticky mucus.
What is CFTR Gene?
Genes are material in our chromosomes containing the code that determine how the cells in the body behave. They provide instructions to create the specific protein the body requires to perform a particular function.
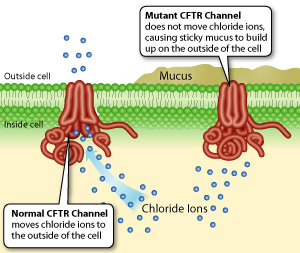
The CFTR gene is a transporter gene located on the 7th chromosome; it provide the information needed to transport sodium and chloride ions across the cell membrane to controls the flow of water in mucus, sweat, tears, saliva and digestive enzyme. When this function fails, too much salt and insufficient water are allowed into the cells.
Cystic fibrosis, like other inherited diseases, is not contagious, nor can it develop in people who have not inherited the faulty gene. Unless someone is born with the disease, they will never have it. However; it is possible for an individual to be a carrier of the defective gene that causes CF.
In normal cells, the CFTR protein acts as a channel for the movement of chloride ions in and out of the cells, a function that is necessary for salt and water balance on epithelial surfaces. If the protein doesn't work correctly and that movement is blocked, an excess of salt and water results in a characteristic build-up of thick, sticky mucus in the tubes and passageways of the body. The build-up of mucus blocks the normal respiratory and digestive function and harbors infection that can cause damage to the lungs, digestive system and other organs, giving rise to symptoms of Cystic fibrosis.
Research suggests, that the CFTR protein also affects the body in other ways and that more than a thousand known abnormalities can affect the CFTR gene. The type of defect a person has can be a factor in the severity of the condition.
Scientists say that the most common mutation (observed in 70% of cystic fibrosis patients) is a three-base deletion in the DNA sequence, causing an absence of a single amino acid in the protein product. Research into this disease continues.
How Cystic Fibrosis Affects Digestion
The digestive system is made up of a group of organs working together to convert food into energy. It breaks down food, absorb the nutrients and excretes the waste product. The pancreas is behind the stomach in the upper abdomen. It secretes digestive enzymes into the small intestine to digest the food we eat.
In people with cystic fibrosis, clogs of thick, viscous secretion prevent the digestive enzymes from getting to the gut, and foods such as fats and proteins cannot adequately digest. The lack of enzymes may cause related symptoms such as large greasy offensive stools. Large stools can often cause irritation, swelling and blockage, making evacuation of the bowel difficult, giving rise to vomiting, bulging of the rectum and collapse of the bowel unto itself. Damage to the bile duct can eventually lead to cirrhosis and liver disease.
Some people with CF may develop distal intestinal obstruction syndrome, an obstruction that occurs in the small bowel causing cramping, constipation and pain.
How Cystic Fibrosis affects nutrition
In babies and children, the lack of nutrition prevents normal growth and development. People of all ages with CF have the common problem of unhealthy weight loss. They find it difficult to gain or maintain a normal weight. Gastroesophageal reflux disease (GERD) is common in people with the condition.
Cystic Fibrosis affect Insulin
In addition to the digestive enzyme, the pancreas also produces the hormone insulin, this helps to control the blood glucose levels. In time, the build up of enzymes unable to reach the small intestine begins to break down the pancreatic tissue that produces insulin (the islets of Langerhans). The loss of tissue can result in slowing down and eventually stopping the production of insulin altogether, resulting in diabetes. Pancreatitis or inflammation of the pancreas can develop due to the blockage of the duct leading from the pancreas to the small intestine.
Cystic Fibrosis Treatment
There is no cure for cystic fibrosis. However, treatments for the condition have much improved over the years. An early diagnosis and a well thought out treatment plan can improve the quality of life and survival rate. Monitoring and follow-up are also important, and where possible, patients with CF should be treated at cystic fibrosis specialty clinics. The aims of CF treatment include:
- Controlling and Preventing Lung Infections, Infection is a frequent occurrence in the lungs of people with cystic fibrosis. Therefore, antibiotics are necessary for the treatment of the condition. Antibiotics can both help to prevent and treat lung and sinus infection. The drugs can be taken by mouth, intravenously or by inhaling through a nebuliser.
- Loosening and Removing the Clogs of Thick Mucus Secretion From the Lungs, airways clearance techniques can help to loosen and rid the lungs of sticky mucus. Removing the mucus can improve lung function and reduce the severity of lung infection. Airway clearance, such as postural drainage and percussion can be done by adult patients themselves, or with help from family, friends and therapists. Electric chest clapper or mechanical percussor are also used to loosen mucus from the lungs.
- Inhaled Medication, there are many types of inhaled medications used to treat CF symptoms. Mucolytics are used to thin the mucus and to easily removed. Antibiotics for the treatment of infections from the bacterium such as Pseudomonas aeruginosin. Hypotonic Saline helps by drawing more water into the airways to loosen the mucus making it easier to remove. Anti-inflammatory medication can contribute to reducing swelling in the airways as a result of infection. Can be taken in the form of inhalation or orally.
- Corticosteroids help to reduce swollen airways to improve breathing, available in sprays and nasal drops.
- Oxygen therapy, this may be given through a face mask or nasal prongs in cases of advanced lung disease.
- Nutrition, as we grow older, our nutritional needs change, to stay healthy, we need the right nutrition. For those with CF, where the intestine cannot absorb vital nutrition, the proper diet is essential. Dietitians at CF centers can offer nutritional programs individually tailored to each stage of life. For growth, development, and the ability to fight off infections, children with CF needs good nutrition and extra calories, there are times when it will be necessary to take extra well-needed calories through a feeding tube. Diets are supplemented with vitamins, A, D, E and K to replace the fat-soluble vitamins that the intestine is unable to absorb. High-calorie shakes will provide extra nutrients, a high salt diet or salt supplements can be taken before exercising, supplemental pancreatic enzymes will help to digest fats and proteins and help to absorb more vitamins.
- Preventing and Treating Blockage in the Intestines, one of the earliest symptoms of bowel obstruction is pain in the right lower abdomen. The pain is often confused with appendicitis since both conditions present with the same symptoms. The absence of enzymes and lack of fluid needed for the digestive process results in constipation that may require intervention. Diet should include foods high in water content. Medication such as enemas, stool softeners and liquids like GoLytely can help to break up the obstruction to better facilitate the evacuation of the bowel. Mucus thinning medication can relieve the blockage. Surgery may be necessary to remove the intestinal obstruction where the blood supply to the intestine has been compromised. Oral medication may be prescribed to reduce stomach acid and to help pancreatic enzymes work more efficiently.
- Preventing dehydration, People with CF don't always feel thirsty even when their bodies are in need of fluids. Dehydration can be dangerous, an individual with CF loses more sodium when ill, or during physical activities than someone without the disease. People with CF are at risk of hyponatraemia dehydration in hot weather. Sodium loss can result in the secretion of aldosterone to cause pseudo-Bartter's syndrome, with hyponatremia ( low level of sodium), hypochloremia ( low chloride iron ), hypokalemia (low potassium) and metabolic alkalosis. Hydration and an increased dietary intake of sodium or supplements are recommended during hot weather. Dehydration in people with CF is best prevented, drinking plenty of fluids and eating salty snacks can help.
- Exercise can help to improve overall fitness, aerobic exercise can loosen and move stubborn mucus as the breathing gets harder, it is beneficial to general health, and can help to keep the bowel moving.
Natural Treatment Include:
- Omega-3 and gamma-linolenic fatty acids, believe to be able to improve pulmonary status
- Omega-3 fatty acids may be able to affect the production of mucus
- Omega-3 fatty acid supplements reduce inflammatory biomarkers like erythrocyte sedimentation rate (ESR) and interleukin-8 concentrations in patients with Cystic Fibrosis
- Ginseng enhances bacterial clearance
- Papaya has been shown to have significant antibacterial action, effective on some bacterium including; Pseudomonas aeruginosin, Staphylococcus aureus, Shigella flexneri
- Licorice is believed to be effective in reducing certain type of pulmonary inflammation
- Bilberry has anti-inflammatory actions
- Fruit and vegetable improve inflammatory markers and oxidative stress in adolescents
- Choline related supplements may help to reduce severity of CF in children
- Coconut oil, studies have shown that coconut oil rich in beneficial saturated fats can relieve conditions such as constipation, acid reflux and GERD. Coconut oil contains antimicrobial properties that can reduce infection and inflammation; this makes it the perfect natural treatment for all intestinal and gastric related problems. Research also show that coconut oil increases vitamin and mineral absorption. For decades, researchers have known that medium-chain fatty acids (MCFAs) in coconut oil were digested differently to other fats, this MCFAs may have significant implications for the treatment of digestive and metabolic conditions. Because the MCFA molecules are smaller than the long chain fatty acids, they require less energy and fewer enzymes to break them down for digestion.
Gene Therapy, With the discovery of the Cystic fibrosis gene in 1989, treatment of the condition was destined to move from the ordinary to the extraordinary. Gene therapy is said to offer the best hope for a life-saving treatment that can get to the very cause of CF, instead of merely treating the symptoms. The concept of gene therapy is to identify the defective gene and to correct the defect with a normal gene.
There are two types of gene therapy. The first, is germ line gene therapy, this therapy, can help not only the person with the condition but also their children. Germ line therapy would change the genetic pool, and future generations would have to live with that change (Genentect Inc., 1998).
The other form of gene therapy is somatic gene therapy (of, or relating to or affecting the body). This therapy involves changing the defective gene in a person, however, in this case, the change will not be inherited by the next generation. This gene therapy is preferred since it does not have the obvious ethical considerations that germ line therapy evokes. According to the Cystic Fibrosis Trust, the first ever trial, looking for clinical benefit following repeated doses of the cf gene wrapped in liposomes, began in 2012.
Patients received gene therapy or a placebo monthly for one year. The trial will last for around 18 months results should be available in 2014.
A breakthrough in stem cell research using mature blood cells from mice is reported by scientists in Japan. The research shows that by shocking blood cells with acid, the scientists were also able to trigger the transformation into stem cells. This new method of reprogramming cells is said to be cheap, simple and fast, with endless possibilities for regenerative medicine without the controversy. Hope for the future?
Just Gene Therapy Lecture : 2012 Trial of Repeated Doses Gene Therapy
New Era In Stem Cell Research
How Cystic Fibrosis Affect the Organs of the Body
How Common is Cystic Fibrosis: Inheriting the disease
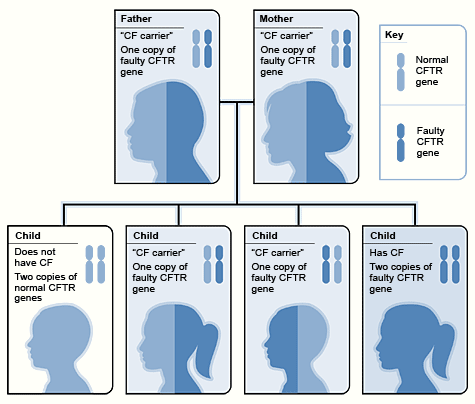
We all inherit two CFTR genes, one from each parents, a child who inherits a defective gene from each parent will have cystic fibrosis.
Someone who inherits one faulty CFTR gene and one normal CFTR gene is a “Cystic Fibrosis carrier.” While a carrier do not usually experience symptoms of the disease, they can pass the defective gene to their children.
Cystic fibrosis is most common in Caucasians of northern European descent. An estimated 1 in 2,500 children born in the UK, are born with Cystic Fibrosis, 1 in 25 are carriers of the disease and around 10,000 people are living with the condition. In the US, 2,500 babies are born with cystic fibrosis each year, about 30,000 people have the disease, more than 10 million Americans carry the cystic fibrosis gene but are not aware of it.
By J Alexis-Hagues © 31/01/2014
Cystic Fibrosis Research: A breath of fresh air
List of genetic disorders include:
Human Disease
| Mutation
| Chromosome
|
|---|---|---|
Angelman Syndrome
| DCP
| 15
|
Color Blindness
| P
| X
|
Down Syndrome
| C
| 21
|
Duchenne Muscular Dystrophy
| D
| Xp
|
Haemochromatosis
| P
| 6
|
Haemophilia A
| P
| X
|
Phenylketonuria
| P
| 12q
|
Polycystic Kidney Disease
| P
| 16 (PKD1) or 4 (PKD2)
|
Prader-Willi Syndrome
| DC
| 15
|
Sickle-cell Disease
| P
| 11p
|
Cystic Fibrosis
| P
| 7q
|
Turner Syndrome
| C
| X
|
Klinefelter Syndrome
| C
| X
|
Diagram shows how two parents who are both CF carriers can pass the defective gene to their children.
Cystic Fibrosis Facts
- Cystic fibrosis is caused by a defective CFTR gene, the gene responsible for making the protein that control the movement of salt and water in and out of the cells.
- CF is one of the most common inherited diseases among Caucasians. 1000 new cases are diagnosed each year
- The symptoms of CF vary over time and from person to person
- Newborn can be screened for CF, diagnosis is based on the results of a number of tests
- Although treatment have greatly improved in recent years, there is no cure for CF
- As treatment continues to improve, life expectancy for people with the disease also improves, currently, some people with CF are living well into their forties, fifties and older
- CF is an inherited disease of secretory glands, such as mucus and sweat glands, pancreas, liver, sinuses, sex organs and intestines
- In people with CF, mucus becomes thick and sticky, it blocks the lungs and airways leading to serious lung infections
- Lung function often begins to worsen in early childhood, causing severe breathing problems.
- CF affects the pancreas by impeding the flow of digestive enzyme, this can prevent the absorption of fats and proteins which can causes vitamin deficiency and malnutrition. Also causes damage to the pancreas affecting the production of insulin resulting in diabetes.
- CF causes excessive loss of salt through sweating, this can result in an imbalance of minerals in the body leading to dehydration, elevated heart rate, tiredness, weakness, low blood pressure, heat stroke and possible death in extreme cases.







