What is Sickle Cell Disease?

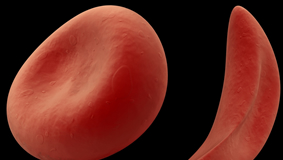
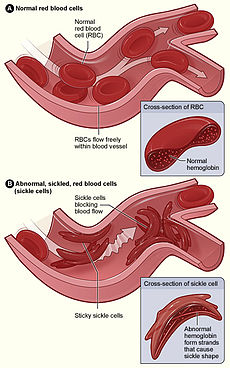
Normal and Sickle Red Blood Cell
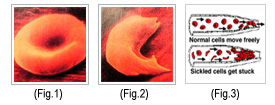
What is Sickle Cell Disease (SCD) or Sickle Cell Anaemia (SCA)?
(Steve Maraboli) Life, the Truth, and Being Free
Sickle cell comes from the Greek word drepanon, meaning sickle as in pruning hook.
Sickle Cell Anemia (SCA) is the most common form of sickle cell disease, also known as Drepanocytosis. Sickle Cell Disease (SCD), is a group of genetic (hereditary) blood disorder that affects the red blood cells. Red blood cells contain a particular protein called hemoglobin (Hb), the function of Hb is to carry oxygen from the lungs to all the different parts of the body. People with Sickle Cell Anemia have what is known as Sickle hemoglobin (HbS) that is different from normal hemoglobin (HbA).
Anemia is a condition where insufficient red blood cells are circulating throughout the body or a deficiency of hemoglobin.
When the sickle hemoglobin releases its oxygen to the tissues, it sticks together to form long rods within the red blood cells and loses its elasticity. The cells become rigid and sickle or crescent shaped instead of the regular biconcave disc-shaped of healthy cells. Consequently; blockage of the small vessels deprives the tissue of oxygen causing a variety of life-threatening complications.
Life expectancy is shortened in people with this inherited genetic disease; studies suggest that in 1994, the average life expectancy for individuals with the condition in the US was estimated to be 42 years in male and 48 years for females. Currently; with improved management, scientific and medical advances, patients are living well into their 70s and beyond.
What causes Sickle Cell Anemia
Sickle cell disease usually present in childhood, it occurs more commonly among people whose ancestors lived in tropical and sub-tropical sub-Saharan regions where malaria is or was once very common.
One-third of all indigenous inhabitants of Sub-Saharan Africa carries the gene. In areas where malaria is prevalent, people carrying a single sickle-cell gene, as in sickle cell trait, are more resistant to malaria. Each year 300,000 African children are born with this genetic disorder, but the condition also exists in North Africa, Greece, Turkey, Saudi Arabia and India.
An estimated 50 million people are affected worldwide, 10,000 individuals in the UK have sickle cell disease.
According to The Centers for Disease Control and Prevention (CDC), although the exact number of people living with Sickle Cell Disease in the US is unknown, an estimated 90,000 to 100,000 American are thought to be affected by SCD. The condition occurs in approximately 1 out of every 500 Black or African-American births, 1 out of every 36,000 Hispanic-American births. Sickle Cell trait (SCT) occurs in around 1 in 12 Black or African-Americans.
While the sickle cell gene occurs in the black population disproportionately, it is not a "black gene," the mutation in the gene that causes sickle cell disease have evolved as resistance to malaria. In areas where malaria is prevalence, the gene is common. When a black person carrying a sickle cell gene has children with a non-black person, the children may inherit the sickle cell gene regardless of race.
The first formal description of sickle cell disease in the Western world was by James Bryan Herrick in 1904.
The Chicago physician noted that blood smear from a 20-year-old male patient from Grenada, who was studying at the Chicago College of Dental Surgery, showed anemia characterized by atypical red cells. Herrick described the shape of the cells as “pear-shaped and elongated forms.” For many years, the disease was called Herrick's Syndrome and changed to “Sickle Cell Disease” by Dr. V.R Mason in 1922.
In 1927, Hahn and Gillespie showed that sickling of the red cells was related to low oxygen.
In 1949, Sickle Cell became the first Molecular disease discovered.
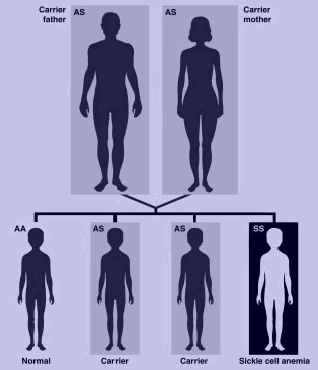
In 1949, geneticist James Neel showed that sickle cell anemia was inherited in recessive Mendelian fashion. (Mendelian inheritance is the manner in which genes and traits are passed from parents to their children)
Sickle cell disease was well known in West Africa under a variety of tribal names that described the symptoms of the disease. Their assessment coincided accurately with current knowledge of the disease.
In 1956, Dr. Vernon M Ingram and J.A. Hunt discovered that SCD was due to a change in a single amino acid in the protein sequence, the replacement of an amino acid in the abnormal hemoglobin.
Ingram and Hunt demonstrated that conversion of glutamic acid to valine at position 6 of the beta-chain was the sole abnormality in sickle hemoglobin. The observation showed that a protein abnormality in which a single amino acid is altered can result in a complex clinical disorder. Today, diseases such as hemophilia and cystic fibrosis are known to arise from an alteration in a single gene. This discovery demonstrated for the first time that genes determine the nature of each amino acid in a protein. In 1978, Tom Maniatis isolated the gene for beta globin.
Sickle Cell Disease results from a genetic mutation that affects the normal development of hemoglobin. A genetic mutation is a permanent change in the usual pattern of genetic information inside all living cells. This shift in pattern leads to one or more of the body's process not working as it should.

What is Sickle Cell trait
We all have two copies of hemoglobin gene, one from each parent. When one of the inherited genes carries the instruction to make sickle haemoglobin and the other carries the instruction to make normal hemoglobin, the person will have Sickle Cell Trait and is a carrier of the sickle cell hemoglobin gene. Someone with sickle cell trait has sufficient normal hemoglobin in their red blood cell to maintain flexibility in the cells and prevent the symptoms of sickle cell disorder. However; they must be careful of situations where there is less oxygen than normal such as taking part in activities at high attitude, scuba diving and when undergoing general anesthetics.
Red blood cells containing HbS are short lived, they have a lifespan of only 10 to 20 days, compared to the healthy red blood cells that last around 120 days. The relatively short life of the HbS cells gives rise to a chronic state of anemia. Nevertheless; people with sickle cell disorder can attend school, college and can work although they will need regular medical attention especially before and after an operation, during pregnancy and when undergoing dental extraction.
The gene for beta globin was isolated by Tom Maniatis in 1978
 comprised of two alpha and two beta globin chains that comes from two multigene cluster.")
Save A Life By Donating Blood and Bone Marrow
Have You Saved A Life Lately? Do You Donate Your Blood/Bone Marrow?
Bone Marrow Donation
Bone marrow is the soft tissue found inside of bones; this is where the immature stem cells are produced. In humans, red blood cells are produced by cores of bone marrow in the head of long bones in a process known as hematopoiesis, a process that produces around 500 billion blood cells per day. Stem cells have the ability to grow into other blood cells such as red blood cells, for carrying oxygen, white cells, to fight infection and platelets for controlling bleeding.
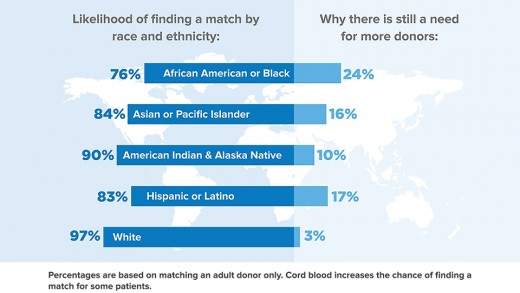
For some patients, the only cure for their condition is a stem cell transplant from a healthy donor. In around 30% of cases, a matching donor can be found within the family from a brother or sister. However; 70% rely on volunteer donors through the Bone Marrow Registry. Finding a suitable match for bone marrow or stem cells transplant is still difficult, and more volunteers are needed to join the register, especially those from ethnic communities.
For people with sickle cell disease, the only known cure is a bone marrow or cord blood transplant, unfortunately; there are serious risks in having a transplant. Therefore, the procedure is only appropriate in patients with severe Sickle cell disease. A transplant replaces the diseased blood-forming cells with healthy cells. The type of transplant used in the treatment of SCD is an allogeneic transplant that uses healthy blood-forming cells from a family member, unrelated donor, or umbilical cord blood unit.
So why is it important to have more volunteers from ethnic communities. A person is more likely to find a donor from the same racial or ethnic background, for matching leukocyte antigen (HLA) types. This is because HLA markers used in matching are inherited. Some combinations of these markers are more common in some racial groups than others. A person's best chance of finding a donor may well be from someone sharing similar ancestry.
The British Bone Marrow Registry (BBMR) is a division of the NHS Blood and Transplant, working in co-operation with the other UK bone marrow/blood donor registry, Anthony Nolan Trust and the NHS Cord blood bank.
There are two ways of donating stem cells, the first, and most frequently used is stem cells from circulation blood. For four days preceding the donation, drugs that can vastly increase the number of stem cells in the donor's circulating blood is given as an injection. On day five, the blood test is taken to check the level of stem cells circulating in the blood. The donor is then connected to a cell separator machine, the machine collects stem cells from the donor's blood through a vein in one arm, and return the blood to the body via a vein in the other arm.
The second method is the donation of the actual bone marrow, this involves the removal of stem cells from the hip bones, the procedure is performed in a hospital under a general anaesthetic using a needle and syringe. Although this is not a surgical operation, the donor may need to remain in the hospital for up to 48 hours and a recovery period at home for approximately five days.
More Donors are needed
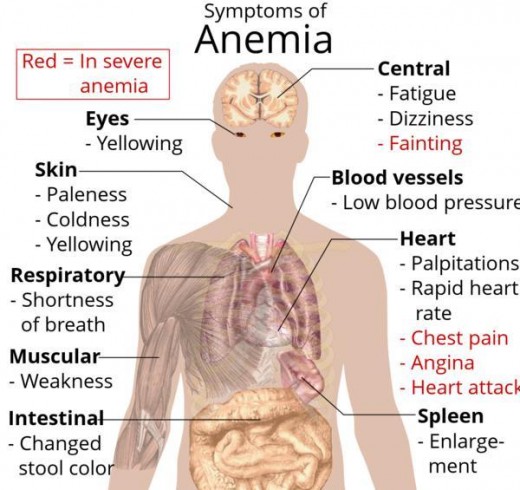
Main Symptoms That May Appear in Anemia
Some Facts about Normal Red blood Cells (RBCs) or Erythrocytes
-
Human RBC takes on average 20 seconds to complete one cycle of circulation
-
Contains special protein known as hemoglobin, an iron-containing biomolecule that can bind oxygen.
-
RBC Survives for an average of 100 to 120 days.
-
About 2 million red blood cells die every second, but the bone marrow produces them just as fast.
-
A normal RBC is biconcave disc-shaped with flattened center
-
RBC begins life as an immature cell in the bone marrow.
-
Controlled by erythropoietin, a hormone produced primarily by the kidneys
-
Unlike most cells, the mature RBC have no nucleus and can easily change shape to flow through small capillaries. No nucleus means more hemoglobin, therefore, more oxygen.
-
One microliter (a single drop) of blood contains: 4.7 to 6.1 million erythrocytes (red blood cells) in male and 4.2 to 5.4 million in female
-
Blood appears red due to the high number of RBC which gets their colour from haemoglobin; oxygenated blood appears bright red and venous blood a darker red
-
The percentage of whole blood volume made up of RBC is known as the haematocrit, the standard measure of RBC
Some Facts about Sickle Red Blood Cells
- Abnormal sickle RBC have a short lifespan; they die after 10 to 20 days.
- Bone marrow cannot make RBC fast enough to replace dying cells, resulting in anemia
- The sickle RBC gets stuck and clogs the blood flow during vaso-occlusive events that can cause infection, pain, stroke and other life-threatening problems.
-
Abnormal sickle cells develop a sickle or crescent shape
-
Sickle cells are stiff and sticky
Sickle Cell Disease; Some Facts and statistics
- People with Sickle Cell Anemia have Sickle hemoglobin (HbS), when sickle hemoglobin gives up its oxygen to the tissue, it sticks together to form long rods within the red blood cells causing them to become rigid and sickle-shaped.
- The different types of Sickle Cell diseases are mainly found in people of Afro-Caribbean descent, Africa, Eastern Mediterranean, Middle East, and Asia.
- In Britain, SDC is most common in people of African and Caribbean descent (1 in 10-40 have sickle cell trait and 1 in 60-200 have SCD)
- 250,000 individuals in the UK are carriers of the sickle cell gene and up to 13,000 people have the sickle disease, making it the most common inherited blood disorder in the country.
- Around 350 babies are diagnosed with the disease every year.
Sickle Cell Test For Newborn Infants
In the UK, all newborn babies are offered a bloodspot test at 5-8 days after birth. The test can help to diagnose medical conditions where early detection can lead to early intervention to make a difference. In Britain, this tests now include screening for SCD.
A small spot of blood is taken from the infant's heel for testing; the result is given to the parents about six weeks later.
Sickle Cell Anemia Symptoms and complications
Sickle cell disease can lead to acute and chronic complications, many of which have a high mortality rate.
Vaso-occlusive Crisis, Sickle Cell Crises also referred to as “sickling crisis” are all terms used to describe several acute conditions that occur in patients with sickle cell disease.
Vaso-occlusive crisis is responsible for a broad range of complication of sickle cell disease, including pain syndrome, leg ulcers, strokes, spontaneous abortions and kidney problems. Acute pain is caused by ischemic tissue injury (the death of tissue resulting from lack of blood supply). Chronic pain occurs due to the destruction of joints and visceral organs from a recurring crisis.
An acute episode of severe pain (crisis) is the primary reason most adolescents and adult patients seek medical intervention, often in hospital emergency departments.
The vaso-occlusive crisis requires collaboration between the family physician and hematologist to deal with acute pain episodes quickly, to effectively manage the long-term chronic pain and to prevent a future crisis that can last between five to seven days.
While it is difficult to predict a vaso-occlusive crisis, there are some precipitating factors such as:
- Dehydration
- Infection
- Cold weather
All signs, symptoms, and complications of SCD are due to two characteristics of red blood cells containing mostly hemoglobin S, the blocking of blood vessels during vaso-occlusive crises, and their very short life span, resulting in chronic anemia.
Chronic anemia (low red blood cells) causes extreme fatigue, shortness of breath, and delayed growth and development in children. The accelerated breakdown of hemoglobin can cause jaundice, and an accumulation of bile stones, most patients with SCD would have had their gallbladder removed by early to middle adulthood. Chronic anemia can eventually lead to life-threatening complication such as congestive heart failure.
Complications of SCD includes:
- Strokes
- Acute Chest Syndrome this is a common complication of SCD, although the pathology is not entirely understood, it is the most common cause of death in patients with SCD. Triggers include infection, vaso-occlusive crisis, and asthma
- Splenic sequestration ( damaged hemoglobin S cells can become trapped in the spleen, associated with a type of life-threatening anemia)
- Recurring infections
- Widespread organ damage
- Pulmonary hypertension, (high blood pressure in the blood vessel supplying the lungs) associated with shortened survival, affects one-third of adult patients with SCD
- Avascular necrosis causes damage to bones, typically affects the arms and legs as result of vaso-occlusion
- Painful and prolonged erections, experienced by at least 30% of boys and men, known as priapism, the condition can lead to impotence
- Psychosocial strain, in addition to pain and brain damage, the condition can also lead to problems in cognition resulting in depression, anxiety, and a feeling of isolation
- Leg ulcers, a result of insufficient blood supply to the skin
- Retinal damage, inadequate blood supply to the retina that can lead to visual impairment and sometimes blindness, some patient also experience bleeding into the retina.
It is imperative that people with SCD understand the risks and seek specialized medical attention immediately if they experience signs and symptoms of any of these serious medical complications.
The information in this article is for educational purposes only; it is not a substitute for medical advice or care.


 Colors Indicate Disease or Health Problems")
")



")
